


一、法律法規(guī)
根據《Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices》(簡稱:《藥機法》),日本藥品和????的管理遵循這一法規(guī)。《藥機法》由原《藥事法》變更而來,其具體實施規(guī)定包括《藥機法實施令》、《QMS省令》以及其他相關公告和通知。
二、主管機關
日本藥品和醫(yī)療器械的管理主要由兩個主管機關負責,即日本厚生勞動省(MHLW)和行政法人醫(yī)藥品醫(yī)療器械綜合機構(PMDA)。
三、醫(yī)療器械分類
(1)一般醫(yī)療器械(ClassⅠ):由日本厚生勞動省大臣在聽取“藥事和食品衛(wèi)生審議會”的意見后進行指定,這類醫(yī)療器械在出現副作用或功能損害時,對人的生命和健康影響的風險較小。
(2)管理醫(yī)療器械(ClassⅡ):同樣由日本厚生勞動省大臣在聽取“藥事和食品衛(wèi)生審議會”的意見后進行指定,這類醫(yī)療器械在出現副作用或功能損害時,有可能對人的生命和健康造成影響,因此需要進行適當的管理。
(3)高度管理的醫(yī)療器械(ClassⅢ、ClassⅣ):這類醫(yī)療器械在出現副作用或功能受損的情況下,會對人的生命和健康產生嚴重影響,因此必須進行嚴格的管理。

四、醫(yī)療器械認證模式和注冊審核流程
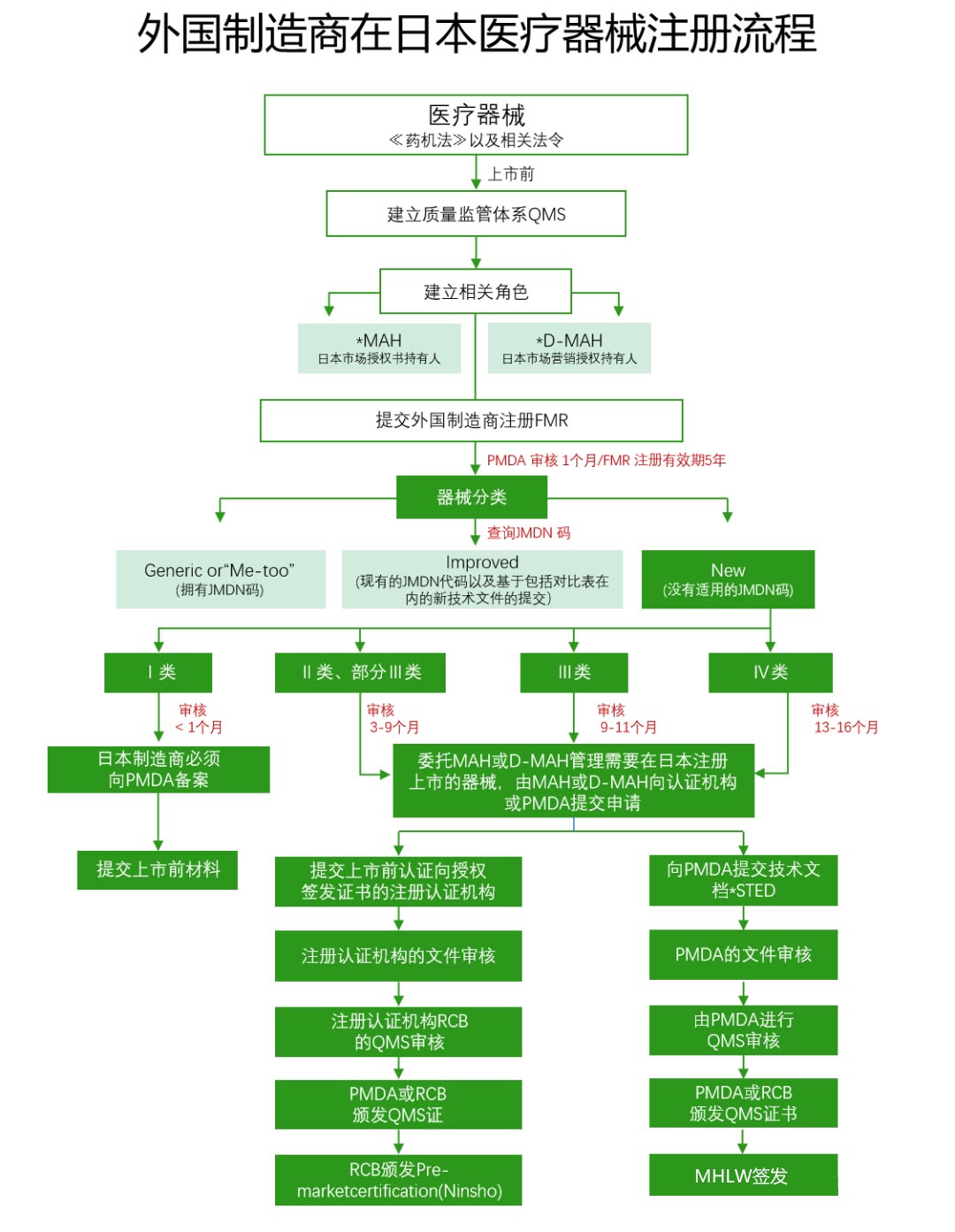
外國制造商如果打算將其設備出口到日本,必須在厚生勞動省進行注冊備案,這個程序被稱為外國制造商注冊(FMR)。這一過程會遵循PMD Act& 第169號法令IS0 13485日本醫(yī)療器械注冊審核流程。
五、重點關注事項
(1)市場授權持有人(MAH)/市場營銷授權持有人(D-MAH):外國制造商必須在日本指定市場授權持有人,這是在日本銷售醫(yī)療器械的首要條件。
(2)STED摘要(注冊資料):需要包括產品規(guī)格、穩(wěn)定性和保質期數據、性能測試數據、風險分析、臨床數據等內容。
(3)質量管理體系(QMS)審核:由醫(yī)藥品與醫(yī)療器械局(PMDA)或某一家注冊過的認證機構(RCB)執(zhí)行,審核范圍包括制造銷售商、醫(yī)療器械的設計、生產制造等所有相關場所。
(4)多法規(guī)要求:進口到日本市場的醫(yī)療器械產品還需要同時滿足其他法規(guī)的要求,如電安法、電波法等。
 ?? ??? WeChat ???
?? ??? WeChat ???